September 28, 2022

Study indicates that ailing neurons may instigate an inflammatory response from the brain’s microglia immune cells
A new study by researchers in The Picower Institute for Learning and Memory at MIT provides evidence from both mouse models and postmortem human tissue of a direct link between two problems that emerge in Alzheimer’s disease: a buildup of double-stranded breaks (DSBs) in the DNA of neurons and the inflammatory behavior of microglia, the brain’s immune cells.
A key new finding is that neurons actively trigger an inflammatory response to their genomic damage. Neurons have not been known to signal the brain’s immune system in Alzheimer’s disease, said study lead author Gwyneth Welch, a former MIT Brain and Cognitive Sciences graduate student in the lab of senior author Li-Huei Tsai.
“This is a novel concept in neuroscience: the idea that neurons can be activating inflammatory activity in response to DNA damage,” Welch said. “The general idea was that neurons have a more passive relationship with microglia regarding age-associated neuroinflammation.”
Instead, what Welch, Tsai and co-authors report in Science Advances is that neurons coping with mounting DSBs go through stages of first trying to fix their fractured DNA and then, when it apparently fails, sending out via molecular signals to microglia, which responded by taking on a more inflammatory state. In experiments where the scientists interrupted the immune signaling, they prevented microglia from entering that state and degrading neural circuit connections, or synapses.
Members of Tsai’s lab have been studying DSBs in the context of Alzheimer’s for more than a decade. Tsai said the new findings add to the emerging understanding of the role they play in Alzheimer’s.
“We have a longstanding interest in understanding DNA breaks in neurons,” said Tsai, Picower Professor of Neuroscience and a founder of MIT’s Aging Brain Initiative. “We previously showed that DNA double stranded breaks are necessary for the induction of activity-regulated gene expression in neurons but we also observed profound DNA damage in neurons in the early stages of neurodegeneration.
“We now know that DNA-damaged neurons exhibit senescent phenotypes and play an active role in eliciting an immune response from microglia and perhaps astrocytes,” Tsai said. “This is mediated by the activation of the NFkappaB transcription factor. Moreover, we identified two cytokines secreted by damaged neurons to recruit microglia and elicit microglial response. Importantly, we show that inhibition of NFkappaB rescued synaptic loss in neurodegeneration, further elucidating of impact of neuroimmune response on synaptic integrity and cognitive function.”
In her thesis research Welch used the lab’s “CK-p25” mouse model of Alzheimer’s, in which disease pathology can be induced, She observed a timeline in which neurons with DSBs appeared within a week, peaked in number after two weeks and then tapered off, becoming notably reduced by six weeks. Meanwhile, those neurons also lost their ability to express a standard marker of neuronal identity. Welch realized there seemed to be stages to the process of coping with DSBs. First neurons have few DSBs and strong identity (baseline), then high DSBs with no loss of identity (stage 1), then high DSBs and a loss of neuronal identity (stage 2).
Transcripts tell the tale
To understand what cells were doing differently in each stage, Welch and the team used multiple “transcriptomics” technologies, which tracks differences in gene expression. Her analyses revealed that neuronal identity genes were most strongly expressed at baseline, DNA repair genes were strong during stage 1, and immune signaling genes were particularly prominent during stage 2.
Among the immune signaling genes were ones governed by the master transcription regulator NFkappaB. These included the cytokines Ccl2 and Cxcl10.
To see if these changes were due specifically to DSBs, Welch treated neurons in the absence of any induced pathology with a chemical called etoposide that causes DSBs. Similar gene expression patterns evident in the induced mice were recapitulated in the ones treated with etoposide. And when Welch also looked at gene expression in the brains of people with DSBs and with Alzheimer’s she also found many significant overlaps.
“We found that stage 1 and stage 2 gene signatures were active in human DSB-bearing neurons,” she and her co-authors wrote. “This neuronal immune signature was further amplified in the context of AD pathology, suggesting that it may serve a functional role in disease-associated neuroinflammation.”
A big role for microglia
Having established that DSB-afflicted neurons employ NFkappaB to send out immune signals such as Ccl2 and Cxcl10, Welch and the team then asked what the effect was. Given that the lab in 2017 had characterized a late-stage inflammatory response on the part of microglia in Alzheimer’s, they hypothesized that neurons might be responsible.
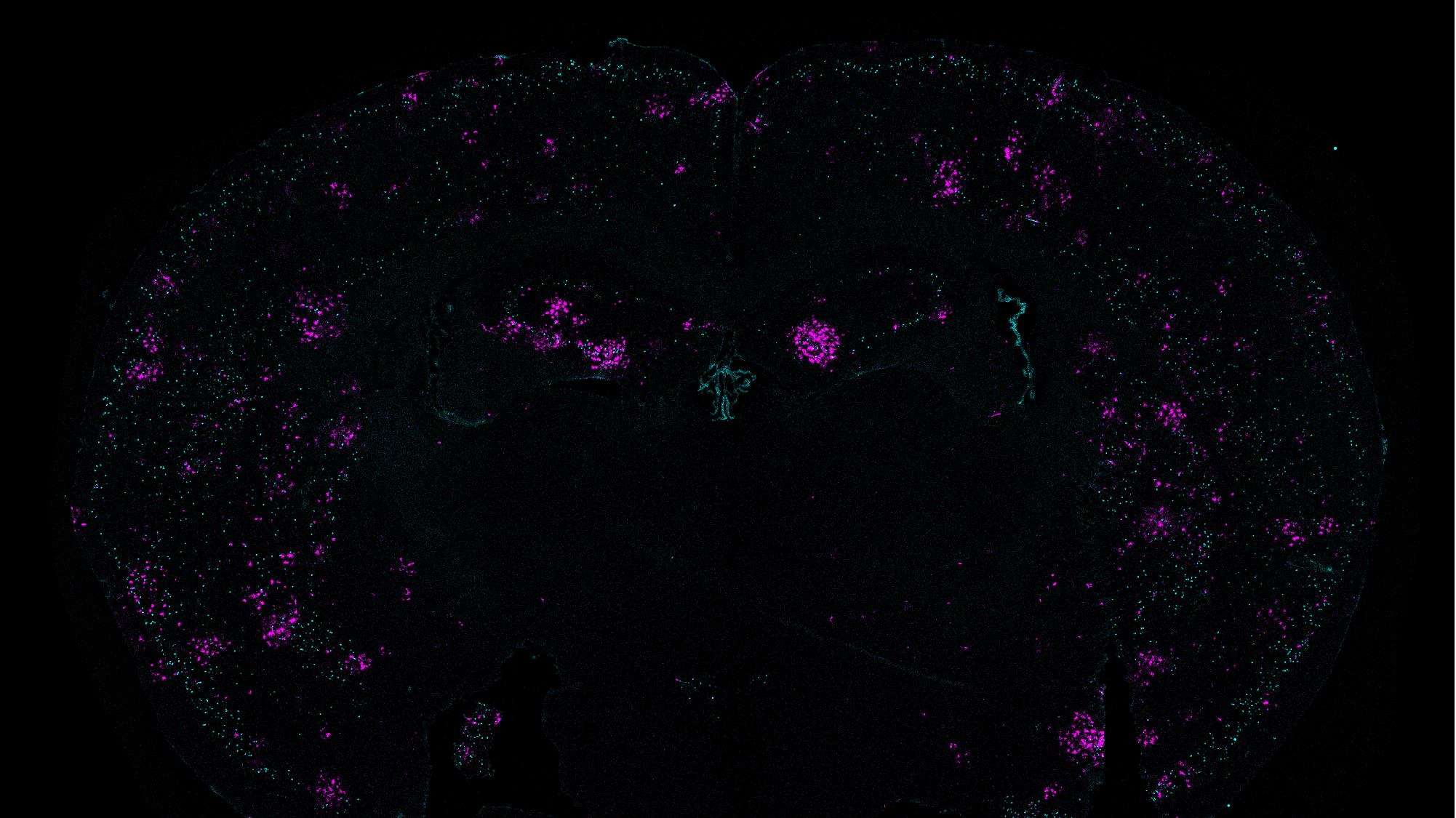
For this analysis Welch used spatial transcriptomics. She divided up both uninduced and induced mouse brains into many areas, and rated each area based on the strength of their DSB signal. Then she analyzed gene transcription in each area and found that locations with high DSBs also had many more microglia in an inflammatory state than locations with low DSBs. They were also able to directly image this relationship, yielding the observation that inflammatory microglia (evidenced by abnormally large cell bodies) were co-located with high-DSB neurons.
To further test the hypothesis, they disrupted NFkappaB regulated transcription in neurons by interfering with a key molecular cog in that machinery called p65. That step resulted in reduced proliferation of microglia and reduced microglia cell body size. It also induced beneficial changes in microglia gene expression, making them more consistent with their normal “homeostatic” state.
In other experiments they found that etoposide-treated neurons expressed Cxcl10 and Ccl2 but that disrupting NFkappaB reduced that expression. They also saw that depleting the two molecules from the brains also prevented microglia from becoming reactive.
And looking back at the neurons, they saw that while knocking down NFkappaB activity didn’t prevent them from dying, it did preserve circuit connections, or synapses, on the neurons that remained alive. That’s important because those circuit connections underlie brain function and microglia are known to prune those.
Because NFkappaB is known to help prevent cell death (which may be why knocking it down didn’t prevent neurons from dying), Welch said knocking it down is not likely to be a therapeutic strategy.
“Its more a proof of principle that that if you turn off a major switch for inflammation, that will change how microglia and neurons interact,” she said. “If your goal is to target inflammatory pathways, focusing on specific signaling molecules might be the more precise way to intervene.”
In addition to Welch and Tsai, the paper’s other authors are Carles Boix, Eloi Schmauch, Jose Davila-Velderrain, Matheus Victor, Vishnu Dileep, P. Lorenzo Bozzelli, Qiao Su, Jemmie D. Cheng, Audrey Lee, Noelle S. Leary, Andreas R. Pfenning, and Manolis Kellis.
The National Institutes of Health, CureAlz, the Glenn Foundation, and the JPB Foundation provided funding for the research.



