
Rett Syndrome, a devastating neurological disorder that typically affects girls, is characterized by stunted growth, impaired language skills, social anxiety and difficulty with movement and breathing. Mutations in the MECP2 gene on the X chromosome (of which girls have two) account for the vast majority of Rett Syndrome cases.
Years of basic research on genes involved in synaptic plasticity led Mriganka Sur’s lab to discover that in mouse models of Rett syndrome, mutations in the gene MECP2 lead to synapse- and circuit-specific problems in the maturation and plasticity of cortical neurons. The dysfunction, which appears to underlie the disorder, arises from a deficit in neuronal signaling molecules that are regulated by MECP2.
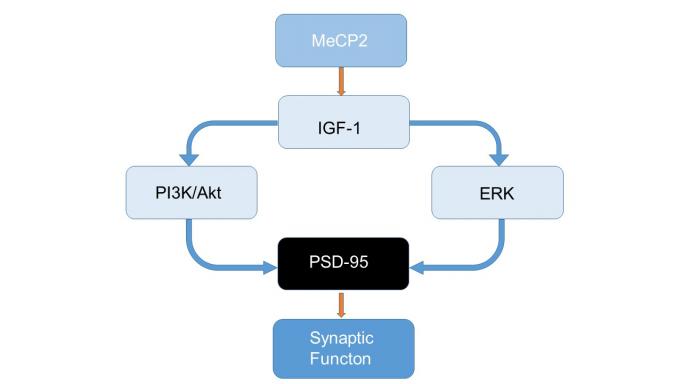
Based on this work, the first mechanism-based pharmacological treatment for Rett Syndrome is now in advanced clinical trials: the molecule IGF1, or Insulin-like growth factor 1. The lab’s hypothesis is that the effects of MeCP2 are mediated in postnatal development through microRNAs that regulate IGF1 signaling. IGF1 is reduced in mouse models of Rett Syndrome.
In a paper in 2014 in the Proceedings of the National Academy of Sciences the team showed that treating mice with the molecule IGF1 corrects functional, structural, and molecular mechanisms downstream of MeCP2 and improves mouse behavior.