March 22, 2024

Single-cell gene expression patterns in the brain’s motor and frontal cortex, and evidence from follow-up experiments, reveal many shared cellular and molecular similarities that could be targeted for potential treatment
On the surface, the movement disorder amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, and the cognitive disorder frontotemporal lobar degeneration (FTLD), which underlies frontotemporal dementia, manifest in very different ways. In addition, they are known to primarily affect very different regions of the brain.
However, doctors and scientists have noted several similarities over the years, and a new study in Cell reveals that the diseases have remarkable overlaps at the cellular and molecular levels, revealing potential targets that could yield therapies applicable to both disorders.
The new paper, led by scientists at MIT and the Mayo Clinic, tracked RNA expression patterns in 620,000 cells spanning 44 different cell types across motor cortex and prefrontal cortex from postmortem brain samples of 73 donors diagnosed with ALS, FTLD, or who were neurologically unaffected.
“We focused on two brain regions that we expected would be differentially affected between the two disorders,” said Manolis Kellis, co-senior author of the paper and professor in the Computer Science and Artificial Intelligence Laboratory at MIT. “It turns out that at the molecular and cellular level, the changes we found were nearly identical in the two disorders, and affected nearly identical subsets of cell types between the two regions.”
Indeed, one of the most prominent findings of the study revealed that in both diseases the most vulnerable neurons were almost identical both in the genes that they express, and in how these genes changed in expression in each disease.
“These similarities were quite striking, suggesting that therapeutics for ALS may also apply to FTLD and vice versa,” said lead corresponding author Myriam Heiman, associate professor in The Picower Institute for Learning and Memory and the Department of Brain and Cognitive Sciences at MIT. “Our study can help guide therapeutic programs that would likely be effective for both diseases.”
Heiman and Kellis collaborated with co-senior author Veronique Belzil, then associate professor of neuroscience at the Mayo Clinic Florida, now director of the ALS Research Center at Vanderbilt University.
“These similarities were quite striking, suggesting that therapeutics for ALS may also apply to FTLD and vice versa. Our study can help guide therapeutic programs that would likely be effective for both diseases.”
-Myriam Heiman, ASSOCIATE PROFESSOR
Another key realization from the study is that brain donors with inherited vs. sporadic forms of the disease showed similarly altered gene expression changes, even though these were previously thought to have different causes. That suggests that similar molecular processes could be going awry downstream of the diseases’ origins.
“The molecular similarity between the familial (monogenic) form and the sporadic (polygenic) forms of these disorders suggests that convergence of diverse etiologies into common pathways,” Kellis said. “This has important implications for both understanding patient heterogeneity and understanding complex and rare disorders more broadly.”
‘Practically indistinguishable’ profiles
The overlap was especially evident, the study found, when looking at the most affected cells. In ALS, known to cause progressive paralysis and ultimately death, the most endangered cells in the brain are upper motor neurons (UMN) in layer 5 of the motor cortex. Meanwhile in behavioral variant frontotemporal dementia (bvFTD), the most common type of FTLD that is characterized instead by changes to personality and behavior, the most vulnerable neurons are spindle neurons, or von Economo cells, found in layer 5 of more frontal brain regions.
The new study shows that while the cells look different under the microscope, and make distinct connections in brain circuits, their gene expression in health and disease is nevertheless strikingly similar.
“UMNs and spindle neurons look nothing alike and live in very different areas of the brain” said Sebastian Pineda, lead author of the study, and a graduate student jointly supervised by Heiman and Kellis. “It was remarkable to see that they appear practically indistinguishable at the molecular level and respond very similarly to disease.”
The researchers found many of the genes involved in the two diseases implicated primary cilia, tiny antenna-like structures on the cell’s surface that sense chemical changes in the cell’s surrounding environment. Cilia are necessary for guiding the growth of axons, or long nerve fibers that neurons extend to connect with other neurons. Cells that are more dependent on this process, typically those with the longest projections, were found to be more vulnerable in each disease.
The analysis also found another type of neuron, which highly expresses the gene SCN4B and which was not previously associated with either disease, also shared many of these same characteristics and showed similar disruptions.
“It may be that changes to this poorly characterized cell population underlie various clinically-relevant disease phenomena,” Heiman said.
The study also found that the most vulnerable cells expressed genes known to be genetically-associated with each disease, providing a potential mechanistic basis for some of these genetic associations. This pattern is not always the case in neurodegenerative conditions, Heiman said. For example, Huntington’s disease is caused by a well-known mutation in the huntingtin gene, but the most highly affected neurons don’t express huntingtin more than other cells, and the same is true for some genes associated with Alzheimer’s disease.
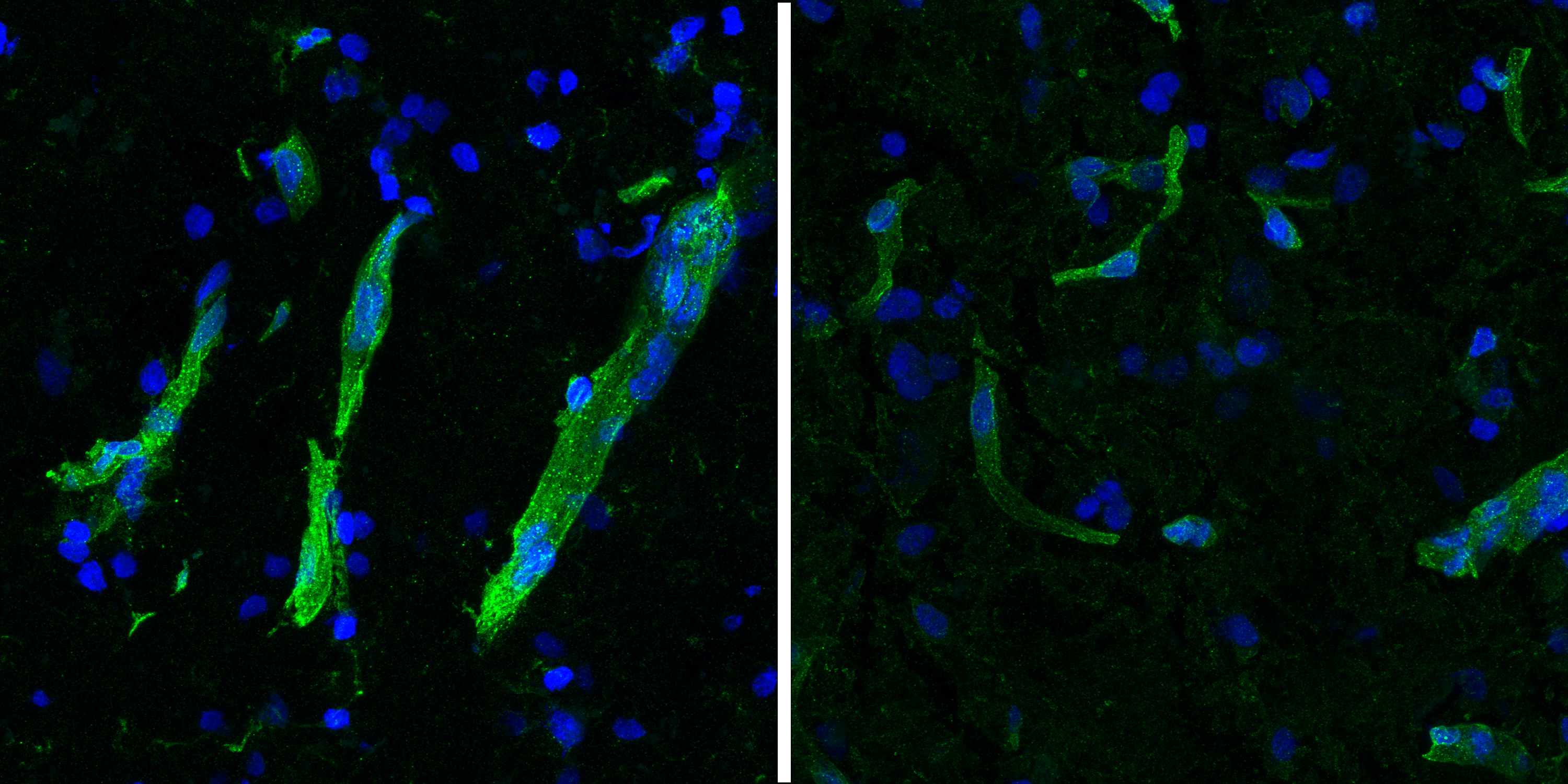
Looking beyond neurons, the study characterized gene expression differences in many other brain cell types. Notably, researchers saw several signs of trouble in the brain’s circulatory system. The blood-brain barrier (BBB), a filtering system that tightly regulates which molecules can go into or come out of the brain through blood vessels, is believed to be compromised in both disorders.
Building on their previous characterization of human brain vasculature and its changes in Huntington’s and Alzheimer’s disease by Heiman, Kellis, and collaborators including Picower Institute Director Li-Huei Tsai, the researchers found that proteins needed to maintain blood vessel integrity are reduced or misplaced in neurodegeneration. They also found a reduction of HLA-E, a molecule thought to inhibit BBB degradation by the immune system.
Given the many molecular and mechanistic similarities in ALS and FTLD, Heiman and Kellis said they are curious why some patients present with ALS and others with FTLD, and others with both but in different orders.
While the present study examined “upper” motor neurons in the brain, Heiman and Kellis are now seeking to also characterize connected “lower” motor neurons in the spinal cord, also in collaboration with Belzil.
“Our single-cell analyses have revealed many shared biological pathways across ALS, FTLD, Huntington’s, Alzheimer’s, vascular dementia, Lewy body dementia, and several other rare neurodegenerative disorders”, says Kellis. “These common hallmarks can pave the path for a new modular approach for precision and personalized therapeutic development, which can bring much-needed new insights and hope.”
In addition to Pineda, Belzil, Kellis and Heiman, the study’s other authors are Hyeseung Lee, Maria Ulloa-Navas, Raleigh Linville, Francisco Garcia, Kyriaktisa Galani, Erica Engelberg-Cook, Monica Castanedes, Brent Fitzwalter, Luc Pregent, Mahammad Gardashli, Michael DeTure, Diana Vera-Garcia, Andre Hucke, Bjorn Oskarsson, Melissa Murray and Dennis Dickson.
Support for the study came from the National Institutes of Health, Mitsubishi Tanabe Pharma Holdings, The JPB Foundation, The Picower Institute for Learning and Memory, the Robert Packard Center for ALS Research at Johns Hopkins, The LiveLikeLou Foundation, the Gerstner Family Foundation, The Mayo Clinic Center for Individualized Medicine, and the Cure Alzheimer’s Fund.



